
Artigo publicado hoje na Nature, uma das mais respeitadas publicações científicas do mundo, descarta a hipótese de que Covid-19 foi criado em laboratório.
Essa tese maluca é difundida por bolsonaristas como se fosse parte de um plano da China para dominar o mundo.
Confira abaixo o artigo:
Desde os primeiros relatos de nova pneumonia (COVID-19) em Wuhan, província de Hubei, China1,2, houve uma discussão considerável sobre a origem do vírus causador, SARS-CoV-23 (também conhecido como HCoV-19) 4 . As infecções por SARS-CoV-2 estão agora disseminadas e, em 11 de março de 2020, 121.564 casos foram confirmados em mais de 110 países, com 4.373 mortes5.
SARS-CoV-2 é o sétimo coronavírus conhecido por infectar seres humanos; SARS-CoV, MERS-CoV e SARS-CoV-2 podem causar doença grave, enquanto HKU1, NL63, OC43 e 229E estão associados a sintomas leves6. Aqui, revisamos o que pode ser deduzido sobre a origem do SARS-CoV-2 a partir da análise comparativa de dados genômicos. Oferecemos uma perspectiva sobre os recursos notáveis do genoma SARS-CoV-2 e discutimos cenários pelos quais eles poderiam ter surgido. Nossas análises mostram claramente que o SARS-CoV-2 não é uma construção de laboratório ou um vírus propositadamente manipulado.
Características notáveis do genoma SARS-CoV-2
Nossa comparação de alfa e betacoronavírus identifica duas características genômicas notáveis do SARS-CoV-2: (i) com base em estudos estruturais7,8,9 e experimentos bioquímicos1,9,10, o SARS-CoV-2 parece ser otimizado para ligação ao receptor humano ACE2; e (ii) a proteína spike de SARS-CoV-2 possui um local de clivagem polibásico (furina) funcional no limite S1 – S2 através da inserção de 12 nucleotídeos8, o que adicionalmente levou à aquisição prevista de três glicanos ligados ao O ao redor do local.
1. Mutações no domínio de ligação ao receptor da SARS-CoV-2
O domínio de ligação ao receptor (RBD) na proteína spike é a parte mais variável do genoma do coronavírus1,2. Demonstrou-se que seis aminoácidos RBD são críticos para a ligação a receptores ACE2 e para determinar a faixa hospedeira de vírus do tipo SARS-CoV7. Com coordenadas baseadas em SARS-CoV, elas são Y442, L472, N479, D480, T487 e Y4911, que correspondem a L455, F486, Q493, S494, N501 e Y505 em SARS-CoV-27. Cinco destes seis resíduos diferem entre SARS-CoV-2 e SARS-CoV (Fig. 1a). Com base em estudos estruturais7,8,9 e experimentos bioquímicos1,9,10, o SARS-CoV-2 parece ter um RBD que se liga com alta afinidade ao ACE2 de humanos, furões, gatos e outras espécies com alta homologia de receptores7.
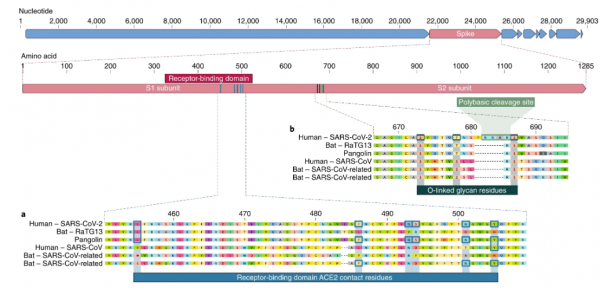
a, Mutações nos resíduos de contato da proteína spike SARS-CoV-2. A proteína spike de SARS-CoV-2 (barra vermelha na parte superior) foi alinhada contra os coronavírus mais semelhantes ao SARS-CoV e o próprio SARS-CoV. Os resíduos principais da proteína spike que fazem contato com o receptor ACE2 são marcados com caixas azuis no SARS-CoV-2 e em vírus relacionados, incluindo o SARS-CoV (cepa Urbani). b, aquisição de sítio de clivagem polibásica e glicanos ligados a O. Tanto o local de clivagem polibásica quanto os três glicanos ligados ao O previstos adjacentes são exclusivos do SARS-CoV-2 e não eram vistos anteriormente nos betacoronavírus da linhagem B. As sequências mostradas são do NCBI GenBank, códigos de acesso MN908947, MN996532, AY278741, KY417146 e MK211376. As seqüências de coronavírus de pangolim são um consenso gerado a partir de SRR10168377 e SRR10168378 (NCBI BioProject PRJNA573298) 29,30.
Imagem em tamanho real
Enquanto as análises acima sugerem que o SARS-CoV-2 pode ligar a ACE2 humana com alta afinidade, as análises computacionais preveem que a interação não é ideal7 e que a sequência RBD é diferente das mostradas no SARS-CoV como sendo ideal para a ligação ao receptor7,11 . Assim, a ligação de alta afinidade da proteína spike SARS-CoV-2 à ACE2 humana é provavelmente o resultado da seleção natural de uma ACE2 humana ou semelhante a humano que permite que outra solução ótima de ligação surja. Esta é uma forte evidência de que o SARS-CoV-2 não é o produto de manipulação intencional.
2. Seleção natural em humanos após transferência zoonótica
É possível que um progenitor de SARS-CoV-2 tenha pulado em seres humanos, adquirindo as características genômicas descritas acima por meio de adaptação durante a transmissão homem-a-homem não detectada. Uma vez adquiridas, essas adaptações permitiriam que a pandemia decolasse e produzisse um conjunto de casos suficientemente grande para acionar o sistema de vigilância que a detectou1,2.
Todos os genomas de SARS-CoV-2 sequenciados até agora têm as características genômicas descritas acima e, portanto, são derivados de um ancestral comum que os possuía também. A presença em pangolins de uma RBD muito semelhante à da SARS-CoV-2 significa que podemos inferir que isso também ocorreu provavelmente no vírus que pulou para os seres humanos. Isso deixa a inserção do local de clivagem polibásica durante a transmissão de homem para homem.
As estimativas do tempo do ancestral comum mais recente da SARS-CoV-2, feito com os dados atuais da sequência, apontam para o surgimento do vírus no final de novembro de 2019 a início de dezembro de 201923, compatível com os primeiros casos confirmados retrospectivamente24. Portanto, esse cenário pressupõe um período de transmissão não reconhecida em humanos entre o evento zoonótico inicial e a aquisição do local de clivagem polibásica. Oportunidades suficientes poderiam ter surgido se houvesse muitos eventos zoonóticos anteriores que produzissem cadeias curtas de transmissão de homem para homem por um período prolongado. Esta é essencialmente a situação do MERS-CoV, para o qual todos os casos humanos são o resultado de repetidos saltos do vírus de camelos dromedários, produzindo infecções únicas ou cadeias de transmissão curtas que acabam por resolver, sem adaptação à transmissão sustentada25.
Estudos de amostras humanas depositadas podem fornecer informações sobre se essa propagação enigmática ocorreu. Estudos sorológicos retrospectivos também podem ser informativos, e alguns estudos foram realizados mostrando exposições de baixo nível a coronavírus do tipo SARS-CoV em certas áreas da China26. Criticamente, no entanto, esses estudos não poderiam ter distinguido se as exposições eram devidas a infecções anteriores com SARS-CoV, SARS-CoV-2 ou outros coronavírus do tipo SARS-CoV. Estudos sorológicos adicionais devem ser conduzidos para determinar a extensão da exposição humana prévia à SARS-CoV-2.
3. Seleção durante a passagem
Pesquisas básicas envolvendo a passagem de coronavírus tipo SARS-CoV de morcego em cultura de células e / ou modelos de animais estão em andamento há muitos anos em laboratórios de nível 2 de biossegurança em todo o mundo27, e existem casos documentados de fugas de laboratório do SARS-CoV28. Portanto, devemos examinar a possibilidade de uma liberação laboratorial inadvertida do SARS-CoV-2.
Em teoria, é possível que o SARS-CoV-2 tenha adquirido mutações RBD (Fig. 1a) durante a adaptação à passagem na cultura de células, como foi observado em estudos de SARS-CoV11. A descoberta de coronavírus do tipo SARS-CoV de pangolins com RBDs quase idênticos, no entanto, fornece uma explicação muito mais forte e parcimoniosa de como o SARS-CoV-2 os adquiriu por recombinação ou mutação19.
A aquisição do local de clivagem polibásica e dos glicanos ligados ao O previstos também argumenta contra cenários baseados em cultura. Novos locais de clivagem polibásica foram observados somente após a passagem prolongada do vírus da influenza aviária de baixa patogenicidade in vitro ou in vivo17. Além disso, uma geração hipotética de SARS-CoV-2 por cultura celular ou passagem de animais exigiria o isolamento prévio de um vírus progenitor com similaridade genética muito alta, o que não foi descrito. A geração subsequente de um local de clivagem polibásica exigiria passagem repetida na cultura de células ou animais com receptores ACE2 semelhantes aos de humanos, mas esse trabalho também não foi descrito anteriormente. Por fim, é improvável que a geração dos glicanos O-preditos tenha ocorrido devido à passagem da cultura celular, pois tais características sugerem o envolvimento de um sistema imunológico18.
Conclusões
No meio da emergência global de saúde pública COVID-19, é razoável imaginar por que as origens da pandemia são importantes. A compreensão detalhada de como um vírus animal ultrapassou os limites das espécies para infectar seres humanos de maneira tão produtiva ajudará na prevenção de futuros eventos zoonóticos. Por exemplo, se o SARS-CoV-2 for pré-adaptado em outra espécie animal, haverá o risco de futuros eventos de reemergência. Por outro lado, se o processo adaptativo ocorreu em seres humanos, mesmo que ocorram transferências zoonóticas repetidas, é improvável que decolem sem a mesma série de mutações. Além disso, a identificação dos parentes virais mais próximos da SARS-CoV-2 circulando em animais ajudará bastante os estudos da função viral. De fato, a disponibilidade da sequência de morcegos RaTG13 ajudou a revelar as principais mutações RBD e o local de clivagem polibásica.
As características genômicas descritas aqui podem explicar em parte a infecciosidade e transmissibilidade da SARS-CoV-2 em humanos. Embora as evidências mostrem que o SARS-CoV-2 não é um vírus propositalmente manipulado, atualmente é impossível provar ou refutar as outras teorias de sua origem descritas aqui. No entanto, uma vez que observamos todos os recursos notáveis do SARS-CoV-2, incluindo o local otimizado de RBD e clivagem polibásica, nos coronavírus relacionados na natureza, não acreditamos que qualquer tipo de cenário laboratorial seja plausível.
Mais dados científicos podem balançar o balanço de evidências para favorecer uma hipótese em detrimento de outra. Obter seqüências virais relacionadas de fontes animais seria a maneira mais definitiva de revelar as origens virais. Por exemplo, uma observação futura de um local de clivagem polibásico intermediário ou totalmente formado em um vírus do tipo SARS-CoV-2 de animais daria ainda mais suporte às hipóteses de seleção natural. Também seria útil obter mais dados genéticos e funcionais sobre SARS-CoV-2, incluindo estudos em animais. A identificação de um potencial hospedeiro intermediário de SARS-CoV-2, bem como o seqüenciamento do vírus em casos muito precoces, também seriam altamente informativos. Independentemente dos mecanismos exatos pelos quais o SARS-CoV-2 se originou por seleção natural, a vigilância contínua de pneumonia em humanos e outros animais é claramente de extrema importância.